")
")
| Issue |
BSGF - Earth Sci. Bull.
Volume 193, 2022
|
|
|---|---|---|
| Article Number | 9 | |
| Number of page(s) | 16 | |
| DOI | https://doi.org/10.1051/bsgf/2022011 | |
| Published online | 04 August 2022 | |
Hexavalent chromium mobility in a high amorphous phase Chromite Ore Processing Residue (COPR) in the perspective of a chromium remediation treatment
Étude de la mobilité du chrome hexavalent dans la perspective d’une dépollution des charrées de chrome à forte teneur en phases amorphes
1
Université de Lille, CNRS, Université Littoral Côte d’Opale, UMR 8187, LOG, Laboratoire d’Océanologie et de Géosciences, 59000 Lille, France
2
Université de Lille, CNRS, Centrale Lille, ENSCL, Université Artois, UMR 8181, Unité de Catalyse et Chimie du Solide, 59000 Lille, France
3
CEREMA – Centre d’études et d’expertise sur les risques, l’environnement, la mobilité et l’aménagement, Direction territoriale Nord-Picardie, 59019 Lille, France
4
DIR-Nord – Direction interdépartementale des routes Nord, Service des politiques et techniques, Cellule Ingénierie entretien chaussées dépendances, 59019 Lille, France
* Corresponding authors: This email address is being protected from spambots. You need JavaScript enabled to view it.
; This email address is being protected from spambots. You need JavaScript enabled to view it.
Received:
23
February
2021
Accepted:
3
June
2022
Abstract
The mineralogical and chemical composition of chromite ore processing residue (COPR) from a site in the north of France (Lille) was investigated. The mineralogical composition was obtained by X-ray diffraction and Rietveld analysis. Geochemical characteristics were established based on elemental analysis, acid leaching, sequential extraction and a chemical equilibrium experiment. Remarkably, this COPR material is composed of 65% silica-rich amorphous phases. Another noticeable result is the presence of about 11% of quartz. Content in toxic Cr(VI) is about 4.9 g/kg, occurring in the solution phase or fixed in unstable crystalline cement phases. Literature data on most studied COPR materials allowed establishing a classification of the materials into (1) high calcium/low silica, (2) intermediate and (3) low calcium/high silica categories. This calcium–silica relation is indicative of the quality of the original ore and the geochemical changes having occurred in a COPR deposit over time, compared with fresh COPR produced from pure ore, and possibly the post-deposit admixture of other waste materials. The Lille material belongs to the third category. The high silica content has influenced the phase associations and their stabilities and favours Cr(VI) mobility. Extraction of leachable Cr(VI) from COPR induces formation of a new chemical equilibrium in the material with a recharge in mobile chromium due to dissolution of cement phases. However, the rate of equilibration is very slow. Four hundred days were needed for the high amorphous phase material in this study. Extraction of leachable Cr(VI) is not a suitable remediation method because it will not allow to withdraw the solid bound Cr(VI) from the material in a single treatment.
Résumé
Cette étude porte sur la caractérisation minéralogique et géochimique des charrées de chrome issues d’un site pollué de la région lilloise dans le nord de la France. La composition minéralogique a été obtenue par diffraction des rayons X et par modélisation Rietveld. La composition géochimique a été déterminée par analyse multi-élémentaire des éléments majeurs et traces du solide, par expérience de dissolution acide, extraction séquentielle et étude de la cinétique de réaction du système charrées de chrome-eau. Une particularité des charrées de chrome étudiées est la très forte teneur en phases minérales cimentaires silicatées amorphes qui s’élève à 65 %. De plus, 11 % des phases minérales cristallisées du matériau sont constituées par le quartz. La teneur en chrome VI est évaluée à 4,9 g/kg, répartie entre la fraction lixiviable et les phases minérales cimentaires cristallisées. Une compilation des données de la bibliographie des charrées de chrome à travers le monde a conduit à une classification des matériaux en trois classes : (1) les charrées de chrome à haute teneur en calcium et basse teneur en silicium, (2) les charrées de chrome à teneur intermédiaire en calcium et en silicium et (3) les charrées de chrome à basse teneur en calcium et haute teneur en silicium. La relation entre le calcium et le silicium est indicatrice de la qualité du minerai de chromite et des modifications géochimiques subies par les charrées lors de l’altération des dépôts. Les charrées de chrome étudiées font partie de la troisième catégorie. La forte teneur en silicium s’exprime à travers une association spécifique des phases minérales dans le matériau et elle semble favoriser la mobilité du Cr(VI). D’autre part, le matériau montre un très lent retour à un état chimique stationnaire après une déstabilisation. Ainsi, un essai d’extraction de fraction lixiviable a induit une dissolution des phases ciments porteuses de Cr(VI) mais environs 400 jours étaient nécessaires pour rétablir un état stable du matériau. À ce stade, les concentrations en Cr(VI) dans l’eau porale étaient revenues à leur niveau initial. L’extraction de la fraction lixiviable du chrome VI n’est pas une solution recommandée de dépollution des charrées de chrome car nécessitant des opérations multiples d’extraction.
Key words: chromite ore processing residue / chromium / waste material / mineralogy / geochemistry / chromium speciation
Mots clés : charrées de chrome / chrome / résidu et déchet industriel / minéralogie / géochimie / spéciation du chrome
© A. Sanchez-Hachair et al., Published by EDP Sciences 2022
 This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
1 Introduction
Chromite ore is mined from igneous-bedded stratiform rocks and ophiolitic deposits (Zubakov and Yusupova, 1962; Edwards and Atkinson, 1986; Gu and Wills, 1988). About 6% of the mined ores are used to satisfy the demands of the chemical industry. Here, chromium is extracted as Cr(VI) following a hydro-metallurgical technique (Supplementary Material S1). The spent ore, called chromium ore processing residue (COPR), is highly enriched in Cr(VI). It has been traditionally deposited as spoil tips on or near the production sites or used as backfill in construction works (Burke et al., 1991; Van Laethem and Legrand, 1993; Bewley, 2007; Kamolpornwijit et al., 2007).
Chromium (VI) is highly toxic for living organisms including humans. It is a carcinogenic and allergenic compound (Norseth, 1981: De Flora et al., 1989: Tennstedt et al., 2012). The COPR material freely deposited on land surfaces or used in construction, represents a serious environmental and health hazard. A large number of studies have been conducted to characterize the speciation of chromium in this material, it’s reactivity and mobility (Geelhoed et al., 2002: Hillier et al., 2003: Kamolpornwijit et al., 2007: Chrysochoou et al., 2009: Elzinga and Cirmo, 2010: Földi et al., 2013: Freese et al., 2014: Matern et al., 2016: Matern and Mansfeldt, 2016: Lehoux et al., 2017). It appears that in COPR, Cr(VI) is not only present as a mobile dissolved or weakly bound phase, but also as a substitute for sulfate in minerals such as ettringite, hydrocalumite and hydrogarnet (Supplementary Material S1). It is important to realise that these phases are not thermodynamically stable and can release Cr(VI) to the material’s pores upon incongruent and congruent dissolution (Geelhoed et al., 2002). Leaching of COPR by surface waters and rain is of environmental concern because of the risk of mobilisation and entrainment of Cr(VI) into non-polluted media. Acid rain can accelerate this process (Xie et al., 2004).
Despite the good solubility of Cr(VI) as chromate (CrO4 2− or HCrO4 −), approaches to remediate the COPR by leaching techniques (pump and treat), passive treatment of solutes (reactive barriers), electrokinetics (electromigration), as well as microbiological methods (bioreduction) have generally failed. The objective of the present study is to highlight the highly dynamic reactions occurring in the material when soluble Cr(VI) is leached out and the time scales of return to equilibrium, which explain the unsuccessful treatments.
The mineralogical and chemical composition of COPR varies significantly from one site to another. Different origins of ore, differences in production processes and the post production history of deposition and aging all influence the mineralogical characteristics of the waste material, the content of chromium and the distribution of chromium among mineral and aqueous phases. The variability of materials will also influence the success of a treatment method. Therefore an effort is made in this study to clearly position the COPR material, which is investigated here, among the many types of materials that have been considered in other studies.
The COPR samples used in the present study originate from a site in the north of France where the material had been used as backfill in infrastructure works. The material is juxtaposed to various inert materials and fly ash. The mineralogical composition of the COPR was determined by Rietveld semi-quantification X-ray powder diffraction method. To recognize the different Cr(VI) reservoirs in the material and to evaluate the lability of Cr(VI) in each of them, the wet chemistry methods of acid leaching and sequential extraction were used. We studied the kinetics of return to chemical equilibrium after initial leaching of the material, with special attention to Cr(VI). These experiments allow to usefully link between the material characterization study and the challenges of material remediation.
2 Materials and methods
2.1 Materials
COPR samples were obtained by drilling into the backfill on site. Each auger provided COPR material, which, back in the laboratory, was precautiously isolated from other materials in the core and mixed for 30 minutes to obtain a well-identified batch of homogeneous stock material. Waste materials such as pieces of vegetation, glass and metallic splinters were removed at this stage. Several drillings were necessary, leading to different batches of material because the amount of COPR extracted from the backfill in one drilling was fairly small (1–2 kg). In this study, we will present results from material PZ2, recovered by an auger drilling in 2013, and from batches of material called T-RX, T-RX2, T-RX3 and T-RX4, recovered from two auger drillings performed in 2015, where COPR occurred in the depth intervals 1.5–3.5 m (T-RX), 1–2 m (T-RX3), 2–3 m (T-RX4) and 3–4 m (T-RX2) [For details see Supplementary Material S2 and Figures S2a and S2b] All drillings were performed within a distance of 1–2 m from each other on the same site. Some analyses could not be conducted in replicate because of material shortage in some experiments.
2.2 Elemental composition of COPR
Elemental composition of COPR samples was determined by Inductively Coupled Plasma Atomic Emission Spectroscopy after lithium borate fusion of the solid samples (SARM–CRPG, Nancy, France). The sample is first molten with lithium borate. After cooling, a solidified bead is recovered, which is subsequently dissolved in acid solution. This solution is then analysed by ICP-AES to determine the major and minor element content of the sample (Al, Ca, Cr, Fe, K, Mg, Mn, Na, Si, S) as well as some trace elements (As, Ba, Pb).
The elemental composition of leachates was also determined by ICP-AES. In this case, a Thermo iCAP6000 SERIES instrument was used (Cerema Hauts-de-France). Aqueous samples were acidified with 1 M nitric acid to reach a pH < 2. Standards were prepared with Merck centipur and Chemlab ICP grade reagents. Samples were handled automatically by a CETAC ASX-520 Autosampler.
2.3 XRPD-Rietveld
Identification and quantification of crystalline phases in COPR was conducted by X-Ray Powder Diffraction (XRPD) method coupled with Rietveld analysis (Rietveld, 1969). The principle of Rietveld refinement is to fit a simulated pattern to the experimental data. Quantitative Rietveld analysis was conducted on samples from T-RX and T-RX4 batches. Detailed description of sample preparation for X-ray analysis can be found in Supplementary Material S3. For T-RX4, the powdered sample was mixed with anatase (TiO2) to determine the ratio between crystalline and amorphous phases in the COPR. For T-RX, the ratio of crystalline to amorphous phases was evaluated by comparing the calcite content as determined by Rietveld analysis and by calcimetry (Bernard calcimeter). X-ray measurements were conducted on a Bruker D4 Endeavor diffractometer at 1.54184 Å Cu-Kα radiation. Phase determination was achieved by reference to patterns published in the scientific literature (Collotti et al., 1959; Smith and Alexander, 1963; Zigan and Rothbauer, 1967; Moore and Taylor, 1970; Sasaki et al., 1979; Winter et al., 1979; Effenberger et al., 1981; Angel, 1988; Sacerdoti and Passaglia, 1988; Hillier et al., 2003; Lenaz et al., 2004; Redhammer et al., 2004). The database Mincryst (Chichagov, 1989) was also used. The above references were selected because the crystallographic information indicated mineral precipitation conditions close to the ones in our material. Quantitative phase analysis was carried out on Jana2006 software (Petricek et al., 2015). For the simulated patterns, we took into account the dominant mineral phases present: ettringite, hydrocalumite, hydrogarnet, quartz, calcite and brucite. The degree of fitting between simulated and experimental diffractograms is expressed through the residual signal.
The Simpson equation (Tallarida and Murray, 1987) for peak area calculation was applied on samples from acid leaching experiments.
2.4 Chromium species quantification in solution and solid phase
Standard methodology for the analysis of aqueous Cr(VI) is by colorimetry of the 1,5 diphenylcarbazide–chromium colour complex (standard method NF T90-093). In a previous study (Sanchez-Hachair and Hofmann, 2018), we proposed an analysis by direct UV-visible spectrophotometry, a method with a better accuracy for samples with high concentrations of Cr(VI) (> 30 mg/L). In the present study, this method was used throughout.
For Cr(VI) quantification in the solid phase, the standard method NF ISO 15192 was applied. Here, the material is first digested under alkaline conditions and then the ionic species in the dissolution brine are separated by an ion chromatography column. This method allows dissolution under redox neutral conditions, ensuring that the oxidation state of chromium species is not altered. The alkaline reagent is obtained by mixing NaOH (0.5 M) with Na2CO3 (0.28 M). An aliquote of 50 mL of this solution is heated at 92.5 °C and 2.5 g of COPR are added. Then, 0.4 g of magnesium chloride and 0.5 mL of phosphate buffer (0.5 M) are added. The sample is stirred for 60 minutes. After chromatographic separation, the Cr(VI) in the alkaline solution is analysed by 1,5 diphenylcarbazide colorimetry or direct spectrophotometry. The analyses were conducted by SGS Multilab Company.
Cr(III) was quantified by taking the difference between total Cr (ICP-AES) and Cr(VI).
2.5 Geochemical experiments
The kinetics of Cr(VI) pore water recharge was investigated. After initial leaching of a COPR sample, the time needed to reach a new equilibrium in Cr(VI) between solid and (pore)solution was determined. This time parameter is essential for the evaluation of a treatment technique. The initial extraction of soluble Cr(VI) was carried out by two successive leaching steps, where alkaline water (pH 11.8 adjusted by NaOH) percolated through the material deposited on a 0.45 μm polycarbonate membrane, pulled by vacuum in a filtration set-up. Water with a pH similar to COPR was used to mimic in situ pH and to prevent dissolution of pH sensitive minerals, in particular the cement phases. This experiment was conducted with the batch T-RX2. An amount of 110 g was percolated with a total of 1.1 L of the alkaline water. Percolation was fast, with a contact time between the material and the water of not more than 10 minutes. The material was then retrieved from the filter and stored in a closed box with a water vapour saturated atmosphere. The material’s evolution was followed over more than one year after the initial extraction. At several times, 5 g samples were retrieved from the box and then submitted to the percolation protocol described above. In the filtrates, the elemental composition was determined by ICP-AES, the Cr(VI) concentration by direct UV-visible spectrophotometry. The chemical composition of the initial leachate is later on referred to as the reference leachate concentration. The pH was measured by a combined pH electrode (Metrohm Unitrode on 781 pH/ion meter). The solid phases were characterized by XRD. The experiment was stopped after 460 days, when the concentrations of dissolved Cr(VI) had reached a new equilibrium. Calculation of the mean kinetic dissolution rate of Cr(VI) is shown in Supplementary Material S7.
Acid leaching experiments can provide information on the dissolution behavior of COPR. In this work, the experimental protocol was designed based on previous work by Geelhoed et al. (2002) and by our group (Lehoux et al., 2017). From a batch of homogenized COPR T-RX2, 15 samples of 10 g were taken and suspended in 50 mL of 1 M NaCl. Then, each sample was adjusted to a different pH, in the range from pH 11.9 to 8.8, by addition of varying amounts of 1 M HCl. The sample pH’es were measured regularly over a period of 27 days and HCl was added as needed to maintain the target pH. We considered that equilibrium was reached when the pH was stable for at least 48 h with no new acid addition. At this stage, solids and solution were separated by vacuum filtration through 0.45 μm polycarbonate filtration membranes. The solid phases remaining after acid treatment were analysed by XRD. The solution phase was analysed for Cr(VI) and major element composition. In Supplementary Material S5, the analytical results are given for the elements Cr, S, Ca, and Mg.
Sequential extraction methods describe the speciation of trace metals in complex materials by identifying the mineral phase reservoirs they are associated with. We followed well-established protocols for sequential extraction, in particular those of Tessier et al. (1979), Zeien and Brummer (1989) and Elzinga and Cirmo (2010), but made modifications to several extraction steps to best take into account the chemical and mineralogical characteristics of COPR (see Supplementary Material S6 for details). Five fractions were defined: the leachable, the exchangeable, the carbonate and cement, the oxide and the residual fractions. A fraction of organically bound Cr(VI) was not included because only traces of organic compounds occur in this material. After each fractionation step, solids and solution were separated by vacuum filtration using 0.45 μm polycarbonate filters. At the end of a step, solids were rinsed with 100 mL de-ionised water to avoid transferring soluble reactive compounds from one step to another. Solutions were analysed for Cr(VI) (colorimetry) and for major and some trace elements (ICP-AES). All reagents were analytical grade. The sequential extraction experiments were conducted on the material batch T-RX. Each sequence of extractions was done with 5 g aliquotes. In the “leachable fraction”, three consecutive leaching tests were performed according to standard NF EN 12457-2 where leaching is conducted with de-ionised water in a solid/solution ratio 1/10. Following the three consecutive tests, the leachates were combined to a unique solution. After the leaching step, the solids were recovered and, during step 2, “the exchangeable fraction”, they were put in contact with 50 mL of 1 M Na2SO4 for 2 h. Sorption studies on ettringite conducted by Myneni (1995) showed that CrO4 2−–SO4 2− exchange only concerns ions adsorbed at the mineral/solution interface while CrO4 2− located within the crystal lattice is not exchangeable. Indeed, ion diffusion in the channel structure of ettringite seems to be very sluggish (Myneni, 1995). However, SO4 2− may desorb CrO4 2− from the LDH lattice structure of hydrocalumite. This LDH is more CrO4 2− selective than SO4 2− (Yamaoka et al., 1989; Yongjun, 2006), but at high sulfate concentrations, ion exchange will proceed nonetheless. Step 3 corresponds to the “carbonate and cement fraction”, in which carbonate minerals as well as cement phases were dissolved. During this step the solids recovered from step 2 were suspended in a solution of 50 mL 1 M ammonium acetate adjusted to pH 6 by 50% acetic acid. Also, this acetic acid preparation is subsequently added to the suspension as needed to maintain the pH constant at 6 over 7 days. The protocol of this step was especially designed to completely dissolve carbonate and cement phases and to neutralize the high acid neutralizing capacity of chromite ore processing residue. Replacement of the “carbonate fraction” used classically in extraction protocols (Tessier et al., 1979; Elzinga and Cirmo, 2010) by a “carbonate and cement fraction” in this work is discussed in more detail in Supplementary Material S6. In the following step, the “oxide fraction” is dissolved. The solids recovered from step 3 are suspended in 62.5 mL of 0.1 mol/L ascorbic acid and 62.5 mL of 0.2 mol/L ammonium oxalate adjusted to pH 3.25 by 50% acetic acid. The extraction is conducted at 96 °C, in the dark for 3 h. Again, the acetic acid preparation is used to maintain the solution pH. The solids recovered from step 4 represent the “residual fraction”. This final fraction is molten by fusion with lithium borate and then taken up in HNO3 acid solution.
3 Results and discussion
3.1 Elemental composition
The elemental composition of the different COPR batches investigated in this study, namely PZ2, T-RX, T-RX2, T-RX3 and T-RX4 is given in Table 1 below. Data are compared to resultants from several other COPR materials.
The elemental composition of COPR has been previously published for different sites. The selection of samples in Table 1 shows that there is a high variability in COPR elemental composition. With regard to chromium, Matern et al. (2016) suggested that COPR with chromium concentrations above 7% should be considered as pure COPR. Materials with lower content may have undergone aging since initial production. The authors explained the chemical difference between COPR materials by four factors: the quality of the chromite ore used for production, the efficiency of chromium extraction during processing of the ore, leaching of COPR by meteoric waters and the mixing of COPR with other waste materials. In this work we propose to describe the “chemical state” of COPR, related to initial ore quality, the history of leaching and contamination by other materials, by its content in calcium and silicon. The COPR data from the Rania and Chiwali sites are taken as a reference. Indeed, the COPR samples were retrieved directly at the metallurgical production site and they have not suffered any alteration prior to analysis (Matern et al., 2016). “Standard COPR” in Figure 1 corresponds to the average composition of these two samples (Rania/Chiwali, Tab. 1).
Silicon is a discriminant element for tracing the history of a COPR because it is generally present at low concentrations in chromite ore. Low Si levels are indicative of good ore quality (Tathavadkar et al., 2003). As discussed above, unusual levels of silicon in COPR may be a marker of material contamination, for instance mixing of COPR with silicate containing waste materials. Calcium is another discriminant element. Although calcium is present in the ore material, the larger part of Ca in COPR comes from the lime that is added to the ore to increase the fusion temperature (Supplementary Material S1). Calcium also allows binding silicate, aluminate and vanadate, which in turn maintains Cr(III) oxidation rates high. Calcium is a major component of amorphous phases such as C–S–H as well as the crystalline cement phases (Supplementary Material S1). Further, it has been shown that hydrated cement phases can loose calcium out of the crystal lattice due to leaching phenomena (Gérard et al., 2002; Harris et al., 2002; Baston et al., 2012). Therefore, low calcium contents can be a marker of COPR leaching. Samples represented in Figure 1 can be grouped into 3 categories, i.e., the high calcium/low silica (COPR standard, Hudson County, Site 7 and Kanpur), the intermediate (Site 4 and Site 2) and the low calcium/high silica (PZ2, T-RX, T-RX2, T-RX3 and T-RX4) materials. “Our” COPR, forms the third group and is characterized by proportionally high silica and low calcium contents. The silicon and calcium parameters help highlighting the fact that COPR materials have a variety of compositions. Comparison of results conducted on different COPR is therefore not straightforward and should be done with precaution.
In the following sections, we will focus on the mineralogy of the COPR series from the Lille site and on the effect of the high silica and low calcium condition on the nature of the cement phases and Cr(VI) mobility.
Elemental composition (%) of COPR in this study and literature data for materials from other sites (major elements (Si, Al, Fe, Ca, Mg, Na, K), minor elements (P, S, Cr), loss of ignition (LOI), and water content %H (in percent)).
Composition élémentaire (%) des charrées de chrome dans cette étude et comparaison avec des données de la littérature.
 |
Fig. 1 Comparison of “our” COPR and COPR from sites published in the literature with “standard COPR”. All data used are given in Table 1. Comparaison des charrées de chrome de cette étude et de charrées d’autres études avec les « charrées de chrome standard ». Les données proviennent du tableau 1. |
3.2 Mineralogical composition and pH stability of phases
X-ray powder diffractograms in Figure 2 show that COPR in this study is composed of quartz, calcite, brucite, chromite, crocoite (PbCrO4), periclase, brownmillerite, hydrogarnet, hydrocalumite, and ettringite. Sporadically anorthite–albite and albite–orthose solid solutions of feldspar may appear in samples.
The diffractograms of all COPR batches show similar mineral composition, however the proportion between mineral occurrences changes from one batch to another. PZ2 is characterized by a much higher ettringite peak and a lower quartz peak than the T-RX materials. In the T-RX series, quartz is present at significant level. The origin of the quartz is questionable. Although low grade ore is known to have been used, in which quartz and silicates were present (Zubakov and Yusupova, 1962; Zhang et al., 2010; Pedrotti, 2012), it was generally avoided because silicate minerals and quartz present in chromite ore will react with sodium carbonate during roasting operation to form a silica gel that hinders oxidation of Cr(III). We cannot exclude at this stage that quartz originating from sources other than the chromite ore was mixed into the COPR after production. In fact, the construction backfill we are investigating is composed of various materials. It is possible that some fly ash or siliceous clay was mixed into the COPR during set-up of the backfill. Visual inspection of our COPR samples by scanning electron microscope and microprobe allows recognizing the presence of quartz particles (Supplementary Material S4; Figs. S4a and S4b). In Figure S4a, individual pure Si grains are observed with 10–40 μm size and irregular, angular shape that certainly correspond to quartz. Figure S4b shows a cut through a silica particle coated with a rim of microsized minerals containing the elements that are typically present in the COPR, namely Ca, Cr, K, Na, Al and P. This object in particular suggests that quartz particles were present in the original ore and that a rim of cement phases was formed around the grains during roasting and subsequent leaching.
For ettringite, hydrocalumite and hydrogarnet, all characteristic XRD peaks are not well-centered on their theoretical position. For ettringite, all our diffractograms show a systematic shift towards higher d values. The maximum shift was observed in sample T-RX3, moving the 9.75 Å d-spacing of ettringite to 10.26 Å. For hydrocalumite we observed an average shift of ± 0.10 Å around the theoretical position of 7.89 Å. The shift for hydrogarnet was also ± 0.10 Å centered on the theoretical position of 5.08 Å. For ettringite, Myneni (1995), who studied sorption and desorption of oxyanions, also observed a significant shift from 9.75 Å to 10.24 Å, which, he postulated, cannot be explained by substitution only because this would require unrealistically high degrees of substitution. As also proposed by Perkins (2000), Myneni (1995) suggested that the peak shift is due to transformation of ettringite into monosulfate and/or monochromate phases at high CrO4/SO4 ratios. This phenomenon may explain why, in the T-RX series of our materials, the ettringite peak at 5.64 Å disappeared (Fig. 2).
While brucite (MgOH)2 was the dominating Mg-mineral in our material, minor quantities of periclase (MgO) were also noted. Thermodynamically, this mineral phase cannot coexist with brucite at high pH (> 10). However, several researchers, studying the mineralogy and texture of COPR, noted the presence of periclase and suggested that this mineral may be protected from hydration when it is located inside nodular structures (Hillier et al., 2003; Matern et al., 2016).
For the batches T-RX and T-RX4, mineral composition was quantified by Rietveld analysis. Only the most important phases of the COPR material, i.e., quartz, calcite, brucite, ettringite, hydrocalumite and hydrogarnet were considered.
Figure 3 shows the residual signals, which arise from the difference between peak intensities of simulated, and experimental diffractograms. When ΔI is positive, the simulated peak is higher than the experimental one, therefore the corresponding phase is overestimated. Our simulations gave rise to overestimated and underestimated mineral phases (Fig. 3). In the case of cement phases, substitution of oxyanions may be responsible for the unsatisfactory fits. Indeed, calculations are made with pure SO4 2− cement phases while minerals in the COPR are partly CrO4 2− substituted. As substitution of SO4 2− by CrO4 2− induces a change in the d-spacing and the peak intensity of the cement phase (Myneni, 1995; Hillier et al., 2007), the standard phases used in calculation are not 100% representative of the phases actually occurring in the material. In addition, in the measured diffractograms, several tiny peaks remained unidentified, suggesting the presence of very minor amounts of other minerals. The low precision of estimates ( Tab. 2) is thought to be due to the sum of these unaccounted signals.
Concerning T-RX4, the sample used in Rietveld analysis did not contain ettringite, while in a second diffractogram made on another sample of this batch of material, the mineral phase was actually identified (Fig. 2). This reveals a great heterogeneity of our COPR even when a same batch of material, coming from the same borehole is considered. These factors of uncertainty must be kept in mind when reading the estimates of mineral composition (Tab. 2).
Table 2 shows the result of Rietveld analysis on samples T-RX and T-RX4. These materials show a high amount of quartz, an unusual result not observed in COPR materials studied by Hillier et al. (2003), Chrysochoou and Dermatas (2007) or Wazne et al. (2008). Occurrence of quartz could be linked to the type of ore used. Another specificity of COPR in the present study is the high content in amorphous phases. We quantified around 65% in T-RX and T-RX4 samples, while Hillier et al. (2003), Wazne et al. (2008), Chrysochoou and Dermatas (2007) only found 30% to 40% of amorphous phases in their materials. During chromium extraction from the ore, a high content in silicate leads to the formation of precursors that will transform to amorphous instead of crystalline phases in the final COPR (see explanations in Supplementary Material S1). Therefore, high silica content and high amorphous phase content in our material may be closely related. This in turn may inversely affect the capacity of the material to fix Cr(VI).
 |
Fig. 2 X-ray powder diffractograms of COPR. The PZ2 diffractogram was acquired over a shorter range of inter reticular spacings d(A) than all other traces. Reticular spacings are presented in logarithmic scale. Et: ettringite; Hc: hydrocalumite; Bw: brownmillerite; Hg: hydrogarnet; Cr: crocoite; Br: brucite; Ch: chromite; Qz: quartz; An: anorthite; Ca: calcite. Periclase, present at minute concentrations, has not been indexed. Diffractogrammes de rayons X des charrées de chrome. |
 |
Fig. 3 Residual signals of Rietveld analysis on T-RX and T-RX4 materials. ΔI is the intensity difference between simulated and experimental XRD traces. ΔI is normalised to 1. Et: ettringite; Hc: hydrocalumite; Bw: brownmillerite; Hg: hydrogarnet; Qz: quartz; Ca: calcite. Signal résiduel de l’analyse de Rietveld des lots T-RX et T-RX4. |
Quantification of crystalline and amorphous mineral phases in COPR batches T-RX and T-RX4. Comparison to data in Hillier et al. (2003). Values in brackets represent standard deviations.
Quantification des phases minérales cristallisées et amorphes dans les charrées de chrome, lots T-RX et T-RX4. Comparaison avec les données dans Hillier et al. (2003).
3.3 Acid leaching experiments
To find out about pH stability of the mineral consortia in our COPR material, acid leaching experiments were carried out with material from batch T-RX2 having an initial pH of 11.9. The experimental pH was lowered to 8.83 in 15 points. The experiment was limited to the pH interval most critical for dissolution and recrystallisation of cement-phases. It is therefore the interval where most Cr(VI) may be released. Figure 4 shows the diffractograms of the original and the final material as well as results from two intermediate pH conditions.
As the diffractograms show, ettringite is most sensitive to pH change. This mineral phase lost 60% between pH 11.9 and pH 11.4 and disappeared entirely at 10.5. Hydrocalumite was also destabilised, with 10% loss at pH 11.4 and 15% at the final pH of 8.83. Calcite and brucite show destabilization similar to hydrocalumite with a loss of 10% at pH 11.4. However, at the final pH calcite and brucite dissolution was stronger and respectively 25% and 20% of these minerals disappeared. Hydrogarnet was not concerned by dissolution over the studied pH range. This is an unexpected result with regard to dissolution studies of other researchers. Geelhoed et al. (2002) observed dissolution of hydrogarnet starting at approximately pH 11 and at pH 9 more than 50% had disappeared (Geelhoed et al., 2002). Theoretical calculations by Geelhoed et al. (2002) even predicted complete dissolution of hydrogarnet below pH 10.8. This difference between theoretical and experimental results points to the important role of the overall composition and texture of the material on the actual stability of the mineral phases.
The difference in mineral dissolution in this study compared to previous is accompanied by another difference concerning the shape of the Cr and S release curves (see Supplementary Material S5). It is observed that below pH 10.5 the two elements do not increase in solution during acid attack as they were expected to do based on results from Geelhoed et al. (2002). We surmise that the sluggish dissolution of hydrocalumite and hydrogarnet and the decreasing trend of Cr and S in increasingly acid solutions is related to some precipitation reaction coupled with inhibition of the expected dissolution of the cement phases, although we do not have direct evidence of secondary mineral phase formation.
The experiments allowed to determine the acid neutralization capacity (ANC) of the samples. For T-RX2, we measured a proton consumption of 0.72 mol H+/kg between pH 11.9 and the experiment end at pH 8.83. While this result agrees with a previous result obtained for PZ2 (Lehoux et al., 2017), it is an order of magnitude lower than the ANC of the material studied by Geelhoed at the same pH edge (6.5 mol H+/kg). This means that the COPR in this study may not guarantee very good long-term pH stability in response to acidity input during meteoric leaching or chemical treatment.
Over all, the mineral phases that were sensitive to dissolution above pH 8.8 comprised ettringite, hydrocalumite, calcite and brucite while hydrogarnet did not react. The following acid sensitivity ladder: ettringite > calcite > brucite > hydrocalumite > hydrogarnet may than be representative for degraded COPR with high silica and high amorphous content. In developing a treatment method for COPR, it will be important to control the input of acidity if mineral phases have to be preserved.
 |
Fig. 4 Diffractograms of T-RX2 samples in the acid leaching experiment. The original material has a pH of 11.9. Anorthite is observed only in the pH 8.83 curve, reflecting random occurrence of feldspar in the batch material. An: anorthite; Et: ettringite; Hc: hydrocalumite; Bw: brownmillerite; Hg: hydrogarnet; Cr: crocoite; Br: brucite; Ch: chromite; Qz: quartz; Ca: calcite. Diffractogrammes d’échantillons T-RX2 de l’expérience de traitement acide. Le pH du matériau initial est de 11,9. |
3.4 Chromium(VI) mobility and extractability
As mentioned previously, Cr(VI) in COPR is easily leached out, contributing to the environmental hazard of the material. However, one time extraction of leachable chromium is not sufficient to remediate the material as has been shown previously in the literature (Antony et al., 2001; Freese et al., 2014; Lehoux et al., 2017; Sanchez-Hachair, 2018). In this section, we first discuss the distribution of Cr(VI) among the different solid phases in the Lille COPR based on sequential extraction experiments and then we investigate reactions and kinetics induced by leaching of the COPR material.
3.5 Sequential extractions
Figure 5 shows the results of the sequential extraction experiments. We will first discuss the mobility of major elements (and some minor and trace elements) in relation with the stepwise dissolution of the mineral phases before investigating the behaviour of chromium.
Least extractable elements are silicon, lead, barium, iron and aluminium, which are present in the “residual fraction” at more than > 85%. These elements belong to mineral phases with low solubility under the extraction conditions, such as quartz, barium chromate and crocoite (PbCrO4). The high content of Al and Fe in the residual fraction is due to stable minerals like brownmillerite, Fe-chromite and possibly some undissolved oxides inherited from the “oxide fraction”. Indeed, dissolution kinetics in COPR is very low (Geelhoed et al., 2002). The elements Cr, K, Na and Ca show intermediate solubility. Their residual fractions are about half of total amounts. The elements S and Mg are the most soluble elements. Residual fractions are as low as 8–20%.
Sulphur and magnesium are mainly bound to the “carbonate and cement fraction” (55% and 75%). Sulphur is extracted as sulfate from the minerals ettringite, hydrocalumite and hydrogarnet while magnesium comes from the mineral brucite. Sulphur is partly leachable (8%) while magnesium release during leaching and exchange steps is almost none on the % scale. During the carbonate and cement step, the pH is lowered to 6, a condition where the surface charge of iron and aluminium oxides shifts to positive values because their points of zero charge (PZC) lie at pH 8–9. Adsorption of anions, in particular sulfate and chromate on Fe- and Al-oxides may thus take place during the carbonate and cement step and be responsible for export of these anions into the oxide fraction. Then, this explains why the COPR extraction data showed 20–25% of sulphur and chromium bound to the oxide fraction.
Calcium is mobilised during the carbonate and cement step (45%) due to calcite and cement phase dissolution. It is not observed in the oxide fraction. Approximately 55% of calcium is bound in high stability phases (residual fraction). An important calcium bearing mineral phase is brownmillerite. Apatite, which is noted frequently in SEM observations, may also contribute to the residual Ca fraction. The fact that calcium is absent from the oxide fraction confirms that carbonate and cement phases have been completely dissolved in their specific extraction step and therefore strengthen the hypothesis that S and Cr in the oxide fraction are due to re-adsorption.
Sodium is mainly associated with two fractions. Around 27% of Na was released from the material during the leaching step, thereby demonstrating the highly labile binding of this element. Sodium was also released to a significant amount in the carbonate and cement step (13%). It is suggested that Na+ is bound to cement phases. For instance, Na-hydrocalumite was recognized in COPR by Chrysochoou et al. (2009). Surprisingly, over half of total sodium was recovered in the residual fraction (55%). Given the high amount of Si in the material, we speculate that Na+ was bound to silicate during the roasting of the ore, leading to the formation of a fairly resistant Na-silicate glass phase.
The COPR contains about 0.6% potassium. Although a small percentage, it is 10–30 times more than in COPR materials from other origins (Tab. 2). Also, in the sequential extractions data, potassium contents carry high standard deviations (Fig. 5). We surmise that potassium is a tracer of external materials mixed into the COPR. The irregular occurrence may be related to the sporadic presence of K-feldspar as suggested by SEM observations.
The study of the distribution of these relevant elements in the sequential fractions allows to get a grip on the actual solubilities of the mineral phases in the material. This will help us understanding the behaviour of chromium. Chromium is observed in all fractions. Beside the already mentioned residual fraction, chromium is present in the “leachable fraction” (> 100 mg/L; 5% of total chromium), in the “carbonate and cement fraction” (15%) and in the “oxide fraction” (20%). Chromium in these three fractions is in the form of Cr(VI) ( Fig. 6). Exchangeable CrO4 2− is very low (< 1%), indicating that it cannot be desorbed neither from the mineral lattice of ettringite (Myneni, 1995), neither from the interlayers of LDH hydrocalumite. Another conclusion to be drawn from the very low exchangeable chromate results is that CrO4 2− adsorption on mineral surfaces is also negligible. Chromium present in the carbonate and cement fraction is interpreted as originating from chromium substituted cement phases. However, during this extraction step, part of the released chromium can be adsorbed on oxide minerals and thereby exported to the oxide fraction. Therefore total chromium bound to cement phases in the original COPR it represented by the sum of chromium in the carbonate and cement, as well as the oxide extraction steps.
It is interesting to compare our results with the work by Elzinga and Cirmo (2010) who studied marsh soils developed on weathered COPR using three different sequential extraction protocols. Their results showed that chromium in the carbonate fraction was less than 10%, while in our study it represented 15% of total chromium. The difference may be explained by the low pH of marsh soil (≈ pH 6), which induces dissolution of chromium binding cement phases. Also, this pH condition is favourable for chromium binding to iron and aluminium oxide phases and indeed Elzinga and Cirmo (2010) determined 50–80% chromium in the oxide fraction, more than the double of our results. This comparison with literature data then clearly illustrates the fact that chromium distribution in COPR will depend to a large extend on the aging history of the material.
We attribute the chromium in the residual fraction (58%) to Cr(III) bound to stable mineral phases such as chromite and brownmillerite (Figs. 5 and 6). In favour of this hypothesis is the fact that no calcium occurred in the oxide fraction, meaning that all Ca-containing cement phases have been dissolved before (during the carbonate and cement step). Then the only chromium left in the material after desorption during the oxide step, must be hardly soluble Cr(III).
Speciation of chromium in the different extraction solutions shows that only Cr(VI) is present in the leachable (1.5 g/kg), exchangeable (0.11 g/kg), carbonate (1.35 g/kg) and oxide fractions (1.96 g/kg). Total Cr(VI) content is then as high as 4.92 g/kg for T-RX sample. This concentration is well comparable to the Cr(VI) content in sample T-RX3 which was determined by alkali digestion (NF ISO 15192) and which also showed 4.92 g/kg. It should be recalled that both batches also showed closely related chemical features (Fig. 1).
Sequential extraction experiments combined with analysis of the chromium oxidation state show that Cr(VI) in COPR is present in a leachable form or bound to easily soluble mineral phases. Only Cr(VI) bound to crocoite (PbCrO4) would be present in the residual fraction. However comparison of Cr(VI) contents in T-RX (sequential extraction) and T-RX3 (alkali digestion) sample suggests that the crocoite fraction is negligible in the overall Cr(VI) balance.
 |
Fig. 5 Distribution of major and some minor and trace elements in the fractions of the sequential extraction experiment conducted on T-RX. Elements are ordered following the trend of increasing residual fraction. Distribution des éléments majeurs, de quelques éléments mineurs et traces dans les différentes fractions de l’expérience d’extraction séquentielle conduite sur T-RX. |
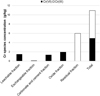 |
Fig. 6 Chromium distribution in the sequential fractions of sample T-RX. Cr(III) was obtained by difference between Cr(total) – ∑Cr(VI). Distribution du chrome dans les fractions de l’expérience d’extraction séquentielle conduite sur T-RX. Le Cr(III) est obtenu par la différence Cr(total) – ∑Cr(VI). |
3.6 Pore solution equilibration experiments
Given the above findings on the distribution of Cr(VI) in the COPR, the pore solution equilibrium experiments were designed to determine the reactivities of the Cr(VI) bearing phases. For this, disequilibrium conditions were created initially and the kinetics of equilibration reactions were followed. As described in the materials section, COPR material was first leached and then, was allowed to equilibrate under humid conditions over a prolonged period of time. New leaching tests were conducted repeatedly, at different aging times, on aliquots of that humid material. Leachates were used to detect the dynamic changes occurring in the pore solution composition over time during equilibration of the material.
The dynamic behaviour of Cr(VI), studied on batch T-RX, is summarised in Figure 7. Leachable Cr(VI), as determined in the sequential extraction study amounted to 1.5 g/kg Cr(VI). Initial leaching in the pore solution equilibration experiment, with only two instead of three rounds of extraction but using de-ionised instead of high pH water, extracted 90% of that amount. Following the initial “cleaning”, ionic strength progressively increased in the pore water until new solid/solution equilibrium was reached after about 400 days of equilibration. At this stage, concentrations of Cr(VI) were back to the initial values as found when untreated COPR material was leached. Cr(VI) initially bound to the solid phase fed the pore solution at a mean kinetic rate of 0.00315 g Cr(VI)/kg/day (see Supplementary Material S7 for details on calculation). This is a remarkable result showing that COPR remediation cannot be achieved by a simple action on the pore water composition, as remediation techniques based on extraction or treatment of pore solutions would do (pressure leaching, electrokinetic leaching; Lehoux et al., 2017). Instead several treatment cycles would be necessary to completely draw Cr(VI) out of the material, which would be strongly time consuming and expensive. In our experiments (Fig. 7), the solid/solution disequilibrium of COPR is obtained by a large flush of water. Under natural conditions, where water percolation will not be as drastic as in our experiments, the time needed for Cr(VI) elimination from COPR by meteoric water leaching may take thousands of years (see Supplementary Material S8 for estimate). Engineering solutions such as reactive barriers intercepting natural flow would need to be supported for immense periods of time. Natural attenuation is also not a treatment solution.
Figure S7 in Supplementary Material S7 gives insight in the reactions taking place in the leached COPR during the long-term equilibration experiment. Besides chromium, solubilisable elements were sulfur, magnesium, calcium, sodium and potassium. Silicon and aluminium were not released to solution. For all these elements, reference leachate concentrations were reached after 400 days equilibration time. However the solubilization steps before attaining these final values were different for the different elements, revealing differences in solid phase speciation and mineral phase dissolution. Four dissolution trends could be identified, schematised in Figure 8.
Trend 1, representative of elements Na, K and Cr(VI) is characterized by two steps, a significant and very fast initial release corresponding to more than 50% of the concentrations in the reference leachate, followed by a steady state which lasted over more than 130 days. Release increased again at longer time, between day 140 and 400, taking the final concentrations to 100% of the reference at 400 days, maintained constant at longer times. Trend 2, representative of S (present as sulfate) is also characterized by a two-step release, but with no steady state phase in between. The reference leachate concentrations are attained at 140 days and no more changes are observed after that maximum. Trend 3, characteristic of Ca, is marked by multiple steps. After an initial fast release (30% C/C0) and a noisy steady state of about 50 days duration, Ca dissolution sets in again, reaching 180 % C/C0 at day 140. This high concentration subsequently decreases since, after day 400, the reference concentration C0 is recorded in the leachate. Calcium and the cations Na and K show opposite solubilization trends, which have similarly been observed by Matern et al. (2020) in a study on COPR dissolution under dynamic conditions. Trend 4 is characteristic of the element Mg. About 10% C/C0 is released initially but this dissolved fraction then progressively vanishes. The principal step of Mg release occurs after day 140, when Mg concentrations are increased to the reference concentrations.
Since the dissolution curves in Figure 8 concern the evolution of the initially leached COPR, it is tempting to attribute the quick rise of concentrations in steps 1 to either the exchangeable or the carbonate and cement fraction of the sequential extraction experiments. Concerning Cr(VI) for which exchangeables were negligible in sequential extraction (Figs. 5 and 6), step 1 may then be directly related to dissolution of cement phases of the COPR. But this process seems be inhibited after some days, when a steady state condition established which we first wrongly interpreted as a final steady state. The inhibitory effect may be due to solute concentrations and pH. But what happened in the material that could explain a revival in dissolution activity after several hundred days of inhibition?
The trend in Ca dissolution (trend 3) may provide a key for understanding. Calcium dissolved above reference concentrations during a step 2 located between 50 and 140 days. At the same time pH in the leachate solutions increased from around 9 in the first days to 11 while the tiny amounts of dissolved Mg disappeared. The step 2 of Cr(VI) release seems to coincide with the decrease in dissolved Ca, a main increase in Mg and a new drop of pH back to 9. Some kind of precipitation of Ca must occur, that entrains a decrease of pH, allowing brucite Mg(OH)2 dissolution and a renewed destabilisation of the cement phases. Our data do not allow to identify the precipitation process. A slow process of carbonation may be invoked with CO2 diffusing from the atmosphere to the sample, causing calcite precipitation and pH drop until at some level brucite dissolution sets in as well as revival of cement dissolution. Another scenario may link calcium decrease to formation of amorphous C–S–H, which also may affect pH and at some stage allow dissolution of brucite and cement. Sulfur, present as sulfate in cement phases is released to equilibrium in two consecutive steps, 140 days were sufficient (trend 2). This reaction does not seem to encounter any inhibition. It can thus be suggested that sulfate release is facilitated relative to chromate release during chemical equilibration of leached COPR.
Release of Cr(VI) from COPR is directly related to the presence in COPR of a leachable phase. The equilibration experiments described above showed that the recharge of this leachable phase after initial purging, is a long lasting process because a large number of solutes as well as secondary reactions intervene in the formation of a new solid/solution steady state.
 |
Fig. 7 Quantification of Cr(VI) fractions in COPR under different conditions. “Equilibrium”, “Disequilibrium” and “New equilibrium” show the Cr(VI) partitioning in T-RX in the untreated material, immediately after initial leaching and 400 days after initial leaching and aging under humid conditions. The “Cr(VI) leachable” fraction of the untreated material corresponds to the leaching step of the sequential extraction experiments, while the “Cr(VI) leachable” of the two other materials was determined as part of the equilibration experiment. UV-Vis spectrophotometry was used in all cases for Cr(VI) quantification. In the COPR after 400 days aging, the “Cr(VI) mineral” was calculated as the difference between the “Cr(VI) mineral” in the untreated COPR and the “Cr(VI) leachable” in the “400 days” sample. Quantification du Cr(VI) dans les charrées de chrome sous les conditions d’« Équilibre », « Déséquilibre » et « Nouvel équilibre », montrant la répartition du Cr(VI) dans le matériau T-RX non traité, immédiatement après le lessivage initial, puis 400 jours après le lessivage initial. |
 |
Fig. 8 Trends in major elements and Cr(VI) concentrations in leachate during the pore solution equilibration experiment. Absolute concentrations C are compared to reference concentration C0 found by initially leaching a fresh sample of COPR using the fast percolation method (see methods and materials). No measurements were made between 140 and 400 days. The lack of data between 140 and 400 days is related to our initially wrong appreciation of kinetic reactions. Indeed, we initially thought that steady state between solid and pore water had been reached after 140 days, because of the observed relative stability of major elemental concentrations (except for calcium and sulfur). Thus after 140 days, we stopped doing measurements. Just out of curiosity, a new measurement was done after 400 days, which revealed that all concentrations had evolved, i.e., that they had levelled off close to their initial values. It can be concluded that 400 days is about the time needed to reach true steady state conditions in the material. The dashed line is drawn by hand and represents a probable course of the curve. Trend 1 is representative of elements Na, K and Cr(VI). Trend 2 is representative of element S, trend 3 of Ca and trend 4 of Mg. The schematic representations are based on chemical data in Figure S7 (Supplementary Material S7). Évolution temporelle des concentrations de quelques éléments majeurs et du Cr(VI) dans les lessivats de l’expérience de ré-équilibration du matériau. |
4 Conclusion
Chromite ore processing residue (COPR) from high lime chromium extraction technique is a highly toxic material putting at risk human health, surface and subsurface environments, and ground waters. Unfortunately, in the past, urban developers have used this material for backfilling in foundations of construction works and for landscaping, because indeed it has good mechanical characteristics and a generally fine grain size in the clay/silt region. With the aim of developing a technique for in situ remediation of this material, we conducted a mineralogical and geochemical study of COPR material originating from a deposit in the area of Lille, north of France. This material showed a high content in silica (7.5% to 18% Si) with quartz present in all samples with up to 10%. The presence of quartz suggests that some mixing of the COPR with other waste materials may have occurred during backfill works, but also that low-grade ore material may have been used in the chromium production. During hydrometallurgical chromium extraction, high silica contents lead to the formation of a silica gel that transforms to amorphous cement phases such as C–S–H and C–A–S–H during cooling and hydration of the COPR. Indeed, the Lille COPR contained 65% of these non-crystalline phases. Thus, the cement mineralogy is strongly impacted by the initial silica content and the Lille COPR differs significantly from COPR materials described in the literature before. A higher fraction of amorphous relative to crystallised cement phases implies that a lower amount of Cr(VI) can be fixed by substitution in the critical cement minerals ettringite, hydrocalumite and hydrogarnet. In other words, a fresh COPR produced from low-grade ore, will initially have a high leachable Cr(VI) content due to a limited Cr(VI) binding mineral reservoir. Over time the Cr(VI) content can then decrease more rapidly so that toxicity may decline relatively faster than in COPR with low amorphous phase content.
When plotting calcium versus silica contents in COPR from various sites, a picture appeared that allows classifying COPRs in high calcium/low silica, intermediate, and low calcium/high silica varieties. We showed that the Lille COPR, which belongs to the low calcium/high silica variety, did not respond to leaching in the same way than COPRs from other localities. During acid leaching down to pH 8.8, only ettringite and hydrocalumite dissolved whereas hydrogarnet did not. This is different from solubilities observed in intermediate COPRs. We also observed inhibitory effects during equilibration of initially leached COPR, leading to stepwise dissolution and long equilibration times probably conditioned by the silica/calcium composition. The proposed classification is a tool to help recognizing the state of chemical evolution of a COPR deposited in the environment. The different degrees of reactivity depending on the COPR category will need thorough investigation in future studies.
In the Lille samples, hexavalent chromium represents about 40% of total chromium. Sequential extraction experiments combined with chromium analysis showed that 70% of total Cr(VI) is present as a substitute anion in cement phases, while 30% are in the leachable fraction. This high content of Cr(VI) in easily soluble phases explains why COPR remediation is difficult. Even when leachable Cr(VI) is extracted from the material, this has been proposed as a remediation solution, COPR will continue producing mobile Cr(VI). Indeed, the cement phases will dissolve and again release Cr(VI) to the pore solution. However chemical equilibration of the destabilised material is a fairly slow process. Under laboratory conditions, 400 days were needed to reach a new steady state between pore water and solid phase after the material had been stripped by leaching. Complex interaction between dissolving cement phases, evolving solutes and formation of new secondary minerals may explain the fairly slow kinetics. For natural attenuation of COPR toxicity, thousands of years would be necessary before this material ceases to release Cr(VI) into the environment. Also, because the pore solution can be recharged with Cr(VI) from the solid phase, a treatment based on one time extraction of leachable Cr(VI) is not sufficient to remediate a COPR deposit.
Supplementary Material
Supplementary Material S1. Formation of Chromite Ore Processing Residue (COPR).
Supplementary Material S2. Sampling of COPR materials.
Supplementary Material S3. XRPD-Rietveld analysis: additional details on methodology.
Supplementary Material S4. Appearance of quartz particles on SEM images.
Supplementary Material S5. Behaviour of elements during acid leaching experiment.
Supplementary Material S6. Specific sequential extraction protocol.
Supplementary Material S7. Pore solution equilibration experiment.
Supplementary Material S8. Estimation of time needed for remediation by natural attenuation.
Access Supplementary MaterialAuthors’ contributions
Arnaud Sanchez-Hachair: Conceptualization, Methodology, Formal analysis and investigation, Writing – original draft preparation, Writing – review and editing. Annette Hofmann: Conceptualization, Writing – original draft preparation, Writing – review and editing, Funding acquisition, Supervision. Guillaume Carlier: Methodology, Materials. Natacha Henry: Formal analysis and investigation, Writing – review and editing. Valentin Bastien: Formal analysis and investigation. Khadijetou Diakite: Formal analysis and investigation. Gaëtan Lefebvre: Writing – review and editing, Materials. Céline Hébrard-Labit: Writing – review and editing, Funding acquisition.
Ethical responsibilities of authors
All authors are aware of the ethical responsibilities, approved and respected COPE guidelines.
Authors’ consent
All authors agreed with the content, agreed to participate and gave explicit consent to submit. The organization, where the work has been carried out, authorised publication of results.
Availability of data and materials
Relevant data are published in the form of tables, histograms or (x,y) graphs within the main text and appendix (Supplementary Material S1–S8).
Conflict of interest
The authors declare that they have no conflict of interest in relation to this article.
Acknowledgement
The French directorate for road infrastructure in the north of France (DIR-Nord) and the French national agency for environment and energy management (ADEME) are acknowledged for funding this research work. Romain Abraham, Marion Delattre and Philippe Recourt (LOG, University of Lille) are thanked for their every day presence and help in the laboratory. We greatly thank Alizée Lehoux who did the first steps in studying the chromite ore-processing residue in our group.
References
- Angel RJ. 1988. High-pressure structure of anorthite. Am. Miner. 73: 1114–1119. [Google Scholar]
- Antony MP, Tathavadkar VD, Calvert CC, Jha A. 2001. The soda-ash roasting of chromite ore processing residue for the reclamation of chromium. Metall. Mater. Trans. B 32: 987–995. [CrossRef] [Google Scholar]
- Baston GMN, Clacher AP, Heath TG, Hunter FMI, Smith V, Swanton SW. 2012. Calcium silicate hydrate (C-S-H) gel dissolution and pH buffering in a cementitious near field. Miner. Mag. 76: 3045–3053. [CrossRef] [Google Scholar]
- Bewley R. 2007. Treatment of chromium contamination and chromite ore processing residue (No. Technical Paper URS Corporation). [Google Scholar]
- Burke T, Fagliano J, Goldoft M, Hazen RE, Iglewicz R, McKee T. 1991. Chromite ore processing residue in Hudson County, New Jersey. Environ. Health Perspect. 92: 131–137. [Google Scholar]
- Chichagov AV. 1989. Information-calculating system on crystal structure data of mineral (MINCRYST). Moscow, USSR. [Google Scholar]
- Chrysochoou M, Dermatas D. 2007. Application of the Rietveld method to assess chromium(VI) speciation in chromite ore processing residue. J. Hazard. Mater. 141: 370–377. [CrossRef] [Google Scholar]
- Chrysochoou M, Fakra SC, Marcus MA, Moon DH, Dermatas D. 2009. Microstructural analyses of Cr(VI) speciation in chromite ore processing residue (COPR). Environ. Sci. Technol. 43: 5461–5466. [CrossRef] [Google Scholar]
- Collotti G, Conti L, Zocchi M. 1959. The structure of the orthorhombic modification of lead chromate PbCrO4 . Acta Crystallogr. 12: 416. [CrossRef] [Google Scholar]
- De Flora S, Camoirano A, Serra D, Bennicelli C. 1989. Genotoxicity and metabolism of chromium compounds. Toxicol. Environ. Chem. 19: 153–160. [CrossRef] [Google Scholar]
- Edwards R, Atkinson K. 1986. Ore deposit geology and its influence on mineral exploration. London (United Kingdom): Ed. Chapman and Hall. [CrossRef] [Google Scholar]
- Effenberger H, Mereiter K, Zemann J. 1981. Crystal structure refinements of magnesite, calcite, rhodochrosite, siderite, smithonite, and dolomite, with discussion of some aspects of the stereochemistry of calcite type carbonates. Z. Für Krist. 156: 233–243. [Google Scholar]
- Elzinga EJ, Cirmo A. 2010. Application of sequential extractions and X-ray absorption spectroscopy to determine the speciation of chromium in Northern New Jersey marsh soils developed in chromite ore processing residue (COPR). J. Hazard. Mater. 183: 145–154. [CrossRef] [Google Scholar]
- Földi C, Dohrmann R, Matern K, Mansfeldt T. 2013. Characterization of chromium-containing wastes and soils affected by the production of chromium tanning agents. J. Soils Sedim. 13: 1170–1179. [CrossRef] [Google Scholar]
- Freese K, Miller R, Cutright T, Senko J. 2014. Review of chromite ore processing residue (COPR): past practices, environmental impact and potential remediation methods. Curr. Environ. Eng. 1: 82–90. [CrossRef] [Google Scholar]
- Geelhoed JS, Meeussen JC, Hillier S, Lumsdon DG, Thomas RP, Farmer JG, et al. 2002. Identification and geochemical modeling of processes controlling leaching of Cr(VI) and other major elements from chromite ore processing residue. Geochim. Cosmochim. Acta 66: 3927–3942. [CrossRef] [Google Scholar]
- Gérard B, Le Bellego C, Bernard O. 2002. Simplified modelling of calcium leaching of concrete in various environments. Mater. Struct. 35: 632–640. [CrossRef] [Google Scholar]
- Gu F, Wills BA. 1988. Chromite – mineralogy and processing. Miner. Eng. 1: 235–240. [CrossRef] [Google Scholar]
- Harris AW, Manning MC, Tearle WM, Tweed CJ. 2002. Testing of models of the dissolution of cements-leaching of synthetic CSH gels. Cem. Concr. Res. 32: 731–746. [CrossRef] [Google Scholar]
- Hillier S, Roe MJ, Geelhoed JS, Fraser AR, Farmer JG, Paterson E. 2003. Role of quantitative mineralogical analysis in the investigation of sites contaminated by chromite ore processing residue. Sci. Total Environ. 308: 195–210. [CrossRef] [Google Scholar]
- Hillier S, Lumsdon DG, Brydson R, Paterson E. 2007. Hydrogarnet: A host phase for Cr(VI) in chromite ore processing residue (COPR) and other high pH wastes. Environ. Sci. Technol. 41: 1921–1927. [CrossRef] [Google Scholar]
- Jacobs M, Tennstedt D, Lachapelle J. 1999. Dermatite allergique de contact. Encycl. Méd. Chir. 7: 1–17. [Google Scholar]
- Kamolpornwijit W, Meegoda Jay N, Zhengbo H. 2007. Characterization of chromite ore processing residue. Pract. Period. Hazard. Toxic Radioact. Waste Manag. 11: 234–239. [CrossRef] [Google Scholar]
- Lehoux AP, Sanchez-Hachair A, Lefebvre G, Carlier G, Hébrard C, Lima AT, et al. 2017. Chromium (VI) retrieval from chromium ore processing residues by electrokinetic treatment. Water. Air. Soil Pollut. 228: 378–391. [CrossRef] [Google Scholar]
- Lenaz D, Skogby H, Princivalle F, Hålenius U. 2004. Structural changes and valence states in the MgCr2O4–FeCr2O4 solid solution series. Phys. Chem. Miner. 31: 633–642. [CrossRef] [Google Scholar]
- Matern K, Mansfeldt T. 2016. Chromate adsorption from chromite ore processing residue eluates by three Indian soils. Environ. Chem. 13: 674. [CrossRef] [Google Scholar]
- Matern K, Kletti H, Mansfeldt T. 2016. Chemical and mineralogical characterization of chromite ore processing residue from two recent Indian disposal sites. Chemosphere 155: 188–195. [CrossRef] [Google Scholar]
- Matern K, Weigand H, Kretschmar R, Mansfeldt T. 2020. Leaching of hexavalent chromium from young chromite ore processing residue. J. Environ. Qual. 49: 712–722. [CrossRef] [Google Scholar]
- Moore AE, Taylor HFW. 1970. Crystal structure of ettringite. Acta Crystallogr. Sect. B 26: 386–393. [CrossRef] [Google Scholar]
- Myneni SCB. 1995. Oxyanion mineral surface interact ions in alkaline environments: AsO4 and CrO4 sorption and desorption in ettringite. Dissertation, The Ohio State University, 265 p. [Google Scholar]
- Norseth T. 1981. The carcinogenicity of chromium. Environ. Health Perspect. 40: 121–130. [CrossRef] [Google Scholar]
- Pedrotti M. 2012. Chromite: from the mineral to the commodity. Dissertation, Università degli studi di Milano, 117 p. [Google Scholar]
- Perkins RB. 2000. The solubility and thermodynamic properties of ettringite, its chromium analogs, and calcium luminum monochromate (3CaO · Al2O3 · CaCrO4 · nH2O). Dissertation, Portland State University, 217 p. [Google Scholar]
- Petricek V, Dusek M, Palatinus L. 2015. Jana2006. Institute of physics. Praha: Academy of sciences of the Czech Republic. [Google Scholar]
- Redhammer GJ, Tippelt G, Roth G, Amthauer G. 2004. Structural variations in the brownmillerite series Ca2(Fe2–xAlx)O5: single-crystal X-ray diffraction at 25 °C and high-temperature X-ray powder diffraction (25 °C ≤ T ≤ 1000 °C). Am. Miner. 89: 405–420. [CrossRef] [Google Scholar]
- Rietveld HM. 1969. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 2: 65–71. [CrossRef] [Google Scholar]
- Sacerdoti M, Passaglia E. 1988. Hydrocalumite from Latium, Italy: its crystal structure and relationship with related synthetic phases. Neues Jahrb. Mineral. Monatshefte 10: 462–475. [Google Scholar]
- Sanchez-Hachair A. (2018). Remediation of polluted soils with chromite ore processing residue (COPR): development of an method coupling electrokinetics and in situ chemical reduction. Confidential dissertation, Université de Lille, 251 p. (in French) [Google Scholar]
- Sanchez-Hachair A, Hofmann A. 2018. Hexavalent chromium quantification in solution: comparing direct UV-visible spectrometry with 1,5-diphenylcarbazide colorimetry. C. R. Chim. 21: 890–896. [CrossRef] [Google Scholar]
- Sasaki S, Fujino K, Takeushi Y. 1979. X-ray determination of electron-density distributions in oxides, MgO, MnO, CoO, and NiO, and atomic scattering factors of their constituent atoms. Proc. Jpn. Acad. Ser. B 55: 43–48. [CrossRef] [Google Scholar]
- Smith GS, Alexander LE. 1963. Refinement of the atomic parameters of α-quartz. Acta Crystallogr. 16: 462–471. [CrossRef] [Google Scholar]
- Tallarida RJ, Murray RB. 1987. Area under a curve: trapezoidal and Simpson ’s rules. In: Manual of Pharmacologic Calculations . New York, NY: Springer, pp. 77–81. [CrossRef] [Google Scholar]
- Tathavadkar VD, Jha A, Antony MP. 2003. The effect of salt-phase composition on the rate of soda-ash roasting of chromite ores. Metall. Mater. Trans. B 34: 555–563. [CrossRef] [Google Scholar]
- Tennstedt D, Jacobs M-C, Baeck M, Lachapelle J-M. 2012. Dermatite allergique de contact. EMC Dermatologie 7(1): 1–17. [CrossRef] [Google Scholar]
- Tessier A, Campbell PG, Bisson M. 1979. Sequential extraction procedure for the speciation of particulate trace metals. Anal. Chem. 51: 844–851. [CrossRef] [Google Scholar]
- Van Laethem F, Legrand J. 1993. Environmental impact of backfill materials of the A22 highway in the north of Lille: search for solutions. Bull. liaison lab. ponts chaussées 188: 59–66. (in French) [Google Scholar]
- Wazne M, Jagupilla S, Moon D, Jagupilla S, Christodoulatos C, Kim M. 2007. Assessment of calcium polysulfide for the remediation of hexavalent chromium in chromite ore processing residue (COPR). J. Hazard. Mater. 143: 620–628. [CrossRef] [Google Scholar]
- Wazne M, Jagupilla SC, Moon DH, Christodoulatos C, Koutsospyros A. 2008. Leaching mechanisms of Cr(VI) from chromite ore processing residue. J. Environ. Qual. 37: 2125. [CrossRef] [Google Scholar]
- Winter JK, Okamura FP, Subrata G. 1979. A high-temperature structural study of high albite, monalbite, and the analbite/monalbite phase transition. Am. Miner. 64: 409–423. [Google Scholar]
- Xie S, Qi L, Zhou D. 2004. Investigation of the effects of acid rain on the deterioration of cement concrete using accelerated tests established in laboratory. Atmos. Environ. 38: 4457–4466. [CrossRef] [Google Scholar]
- Yamaoka T, Abe M, Tsuji M, 1989. Synthesis of Cu–Al hydrotalcite like compound and its ion exchange property. Mater. Res. Bull. 24: 1183–1199. [CrossRef] [Google Scholar]
- Yongjun F. 2006. Formation et propriétés de materiaux hydroxydes doubles lamellaires bi-intercalés. Agrégats moléculaires et atomiques. English NNT: 2006CLF21708. Dissertation, Université Blaise Pascal–Clermont-Ferrand II. [Google Scholar]
- Zeien H, Brummer GW. 1989. Chemische Extraktion zur Bestimmung von Schwermetallbindungsformen in Boden. Mitteilungen der Deutschen Bodenkundlichen Gesellschaft 59: 505–510. [Google Scholar]
- Zhang Y, Zheng S, Du H, Xu H, Zhang Y. 2010. Effect of mechanical activation on alkali leaching of chromite ore. Trans. Nonferrous Met. Soc. China 20: 888–891. [CrossRef] [Google Scholar]
- Zigan F, Rothbauer R. 1967. Neutronenbeugungsmessungen am Brucit. Neues Jahrb. Miner. Monatshefte 1967: 137–143. [Google Scholar]
- Zubakov SM, Yusupova EN. 1962. Composition and properties of chromite ores from new deposits in Kazakhstan. Refractories 3: 341–344. [CrossRef] [Google Scholar]
Cite this article as: Sanchez-Hachair A, Henry N, Bastien V, Diakite K, Carlier G, Lefebvre G, Hébrard-Labit C, Hofmann A. 2022. Hexavalent chromium mobility in a high amorphous phase Chromite Ore Processing Residue (COPR) in the perspective of a chromium remediation treatment, BSGF - Earth Sciences Bulletin 193: 9.
All Tables
Elemental composition (%) of COPR in this study and literature data for materials from other sites (major elements (Si, Al, Fe, Ca, Mg, Na, K), minor elements (P, S, Cr), loss of ignition (LOI), and water content %H (in percent)).
Composition élémentaire (%) des charrées de chrome dans cette étude et comparaison avec des données de la littérature.
Quantification of crystalline and amorphous mineral phases in COPR batches T-RX and T-RX4. Comparison to data in Hillier et al. (2003). Values in brackets represent standard deviations.
Quantification des phases minérales cristallisées et amorphes dans les charrées de chrome, lots T-RX et T-RX4. Comparaison avec les données dans Hillier et al. (2003).
All Figures
|
Fig. 1 Comparison of “our” COPR and COPR from sites published in the literature with “standard COPR”. All data used are given in Table 1. Comparaison des charrées de chrome de cette étude et de charrées d’autres études avec les « charrées de chrome standard ». Les données proviennent du tableau 1. |
| In the text | |
|
Fig. 2 X-ray powder diffractograms of COPR. The PZ2 diffractogram was acquired over a shorter range of inter reticular spacings d(A) than all other traces. Reticular spacings are presented in logarithmic scale. Et: ettringite; Hc: hydrocalumite; Bw: brownmillerite; Hg: hydrogarnet; Cr: crocoite; Br: brucite; Ch: chromite; Qz: quartz; An: anorthite; Ca: calcite. Periclase, present at minute concentrations, has not been indexed. Diffractogrammes de rayons X des charrées de chrome. |
| In the text | |
|
Fig. 3 Residual signals of Rietveld analysis on T-RX and T-RX4 materials. ΔI is the intensity difference between simulated and experimental XRD traces. ΔI is normalised to 1. Et: ettringite; Hc: hydrocalumite; Bw: brownmillerite; Hg: hydrogarnet; Qz: quartz; Ca: calcite. Signal résiduel de l’analyse de Rietveld des lots T-RX et T-RX4. |
| In the text | |
|
Fig. 4 Diffractograms of T-RX2 samples in the acid leaching experiment. The original material has a pH of 11.9. Anorthite is observed only in the pH 8.83 curve, reflecting random occurrence of feldspar in the batch material. An: anorthite; Et: ettringite; Hc: hydrocalumite; Bw: brownmillerite; Hg: hydrogarnet; Cr: crocoite; Br: brucite; Ch: chromite; Qz: quartz; Ca: calcite. Diffractogrammes d’échantillons T-RX2 de l’expérience de traitement acide. Le pH du matériau initial est de 11,9. |
| In the text | |
|
Fig. 5 Distribution of major and some minor and trace elements in the fractions of the sequential extraction experiment conducted on T-RX. Elements are ordered following the trend of increasing residual fraction. Distribution des éléments majeurs, de quelques éléments mineurs et traces dans les différentes fractions de l’expérience d’extraction séquentielle conduite sur T-RX. |
| In the text | |
|
Fig. 6 Chromium distribution in the sequential fractions of sample T-RX. Cr(III) was obtained by difference between Cr(total) – ∑Cr(VI). Distribution du chrome dans les fractions de l’expérience d’extraction séquentielle conduite sur T-RX. Le Cr(III) est obtenu par la différence Cr(total) – ∑Cr(VI). |
| In the text | |
|
Fig. 7 Quantification of Cr(VI) fractions in COPR under different conditions. “Equilibrium”, “Disequilibrium” and “New equilibrium” show the Cr(VI) partitioning in T-RX in the untreated material, immediately after initial leaching and 400 days after initial leaching and aging under humid conditions. The “Cr(VI) leachable” fraction of the untreated material corresponds to the leaching step of the sequential extraction experiments, while the “Cr(VI) leachable” of the two other materials was determined as part of the equilibration experiment. UV-Vis spectrophotometry was used in all cases for Cr(VI) quantification. In the COPR after 400 days aging, the “Cr(VI) mineral” was calculated as the difference between the “Cr(VI) mineral” in the untreated COPR and the “Cr(VI) leachable” in the “400 days” sample. Quantification du Cr(VI) dans les charrées de chrome sous les conditions d’« Équilibre », « Déséquilibre » et « Nouvel équilibre », montrant la répartition du Cr(VI) dans le matériau T-RX non traité, immédiatement après le lessivage initial, puis 400 jours après le lessivage initial. |
| In the text | |
|
Fig. 8 Trends in major elements and Cr(VI) concentrations in leachate during the pore solution equilibration experiment. Absolute concentrations C are compared to reference concentration C0 found by initially leaching a fresh sample of COPR using the fast percolation method (see methods and materials). No measurements were made between 140 and 400 days. The lack of data between 140 and 400 days is related to our initially wrong appreciation of kinetic reactions. Indeed, we initially thought that steady state between solid and pore water had been reached after 140 days, because of the observed relative stability of major elemental concentrations (except for calcium and sulfur). Thus after 140 days, we stopped doing measurements. Just out of curiosity, a new measurement was done after 400 days, which revealed that all concentrations had evolved, i.e., that they had levelled off close to their initial values. It can be concluded that 400 days is about the time needed to reach true steady state conditions in the material. The dashed line is drawn by hand and represents a probable course of the curve. Trend 1 is representative of elements Na, K and Cr(VI). Trend 2 is representative of element S, trend 3 of Ca and trend 4 of Mg. The schematic representations are based on chemical data in Figure S7 (Supplementary Material S7). Évolution temporelle des concentrations de quelques éléments majeurs et du Cr(VI) dans les lessivats de l’expérience de ré-équilibration du matériau. |
| In the text | |
Current usage metrics show cumulative count of Article Views (full-text article views including HTML views, PDF and ePub downloads, according to the available data) and Abstracts Views on Vision4Press platform.
Data correspond to usage on the plateform after 2015. The current usage metrics is available 48-96 hours after online publication and is updated daily on week days.
Initial download of the metrics may take a while.